"... the powerhouse of the cell, epitomize the intricate dance of energy production and cellular resilience, embodying nature's ingenious design in every pulsating breath of life."
Mitochondria, often referred as the "powerhouses of the cell," stand as dynamic organelles essential for cellular energetics and metabolic homeostasis. These multifaceted structures, with their intricate membranes and matrix compartments, orchestrate a myriad of biological processes critical for cellular function and the survival of the organism. From energy production to calcium homeostasis, apoptosis regulation to reactive oxygen species (ROS) management, mitochondria serve as molecular hubs orchestrating a plethora of physiological functions.
Mitochondria Function
At the core of mitochondria's functionality lies oxidative phosphorylation (OXPHOS), a finely tuned biochemical process catalyzing the conversion of nutrients into adenosine triphosphate (ATP), the universal energy currency of the cell. Through a series of enzymatic reactions orchestrated within the electron transport chain (ETC), electrons derived from metabolic substrates are shuttled across mitochondrial inner membranes, driving proton translocation and establishing an electrochemical gradient. This proton motive force, harnessed by ATP synthase, fuels the synthesis of ATP from adenosine diphosphate (ADP) and inorganic phosphate, culminating in cellular energy provision.
Beyond ATP synthesis, mitochondria serve as signaling platforms integrating metabolic cues with cellular responses. Mitochondrial dynamics, encompassing fission, fusion, and mitophagy, regulate organelle morphology and function, dictating cellular adaptation to changing metabolic demands and stress stimuli. Moreover, mitochondria interface with various cellular pathways, including calcium signaling, redox homeostasis, and apoptosis, exerting profound effects on cell fate and survival.
Mitochondria Made Easy: A Simple Overview for Beginners
Mitochondria are like tiny power stations inside our cells. They play a crucial role in keeping our bodies running smoothly by producing energy that cells need to function properly. Think of them as tiny batteries that power everything our cells do, from moving and growing to repairing damage.
Mitochondria use a process called oxidative phosphorylation to turn nutrients from food into a form of energy called ATP. This energy is like fuel for our cells, helping them carry out all their jobs. Mitochondria also help regulate other important processes in the cell, like controlling levels of calcium and deciding when cells should die.
Sometimes, mitochondria can't do their job properly due to genetic problems, toxins in the environment, or just aging. This can lead to what scientists call "mitochondrial dysfunction." When mitochondria aren't working as they should, cells can't get enough energy, and other problems can occur. This can affect many parts of the body, including the brain and nervous system.
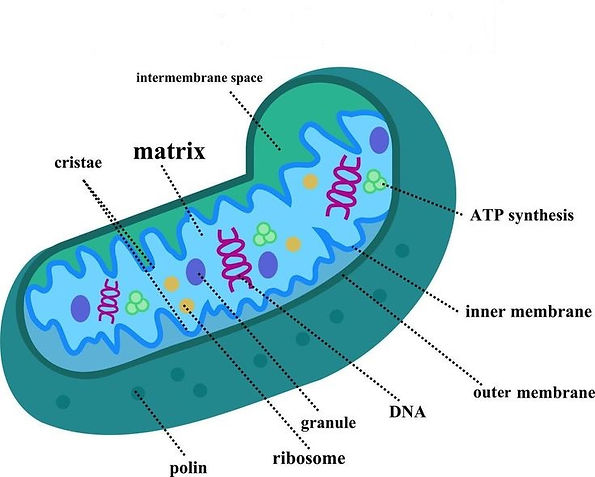
Mitochondrial Dysfunction
Perturbations in mitochondrial function precipitate a spectrum of pathological sequelae, ranging from metabolic disorders to neurodegenerative diseases. Mitochondrial dysfunction manifests through diverse mechanisms, encompassing genetic mutations, environmental insults, and aging-related decline. Impaired oxidative phosphorylation compromises ATP production, culminating in bioenergetic insufficiency and cellular dysfunction. Concurrently, dysregulated ROS production and mitochondrial calcium overload inflict oxidative damage and trigger apoptotic cascades, exacerbating cellular demise.
Mitochondrial dysfunction extends beyond energetic deficits, permeating diverse cellular processes and organ systems. Neurological tissues, with their high-energy demands and susceptibility to oxidative stress, are particularly vulnerable to mitochondrial impairments. Mitochondrial disorders, characterized by genetic aberrations affecting mitochondrial DNA (mtDNA) or nuclear-encoded mitochondrial proteins, underlie a constellation of clinical phenotypes, encompassing Leigh syndrome, mitochondrial encephalomyopathy, and neurodegenerative disorders like Parkinson's and Alzheimer's diseases.
Among the diverse array of conditions associated with mitochondrial dysfunction, emerging research has shed light on its intriguing connection to autism spectrum disorder (ASD) and other neurodevelopmental disorders. Recent studies have demonstrated mitochondrial abnormalities and impaired oxidative phosphorylation in individuals with ASD, suggesting a potential mechanistic link between mitochondrial dysfunction and the pathophysiology of autism (Goh et al., 2021; Rose et al., 2020). Furthermore, mitochondrial dysfunction has been implicated in a spectrum of neurodevelopmental disorders beyond autism, including Rett syndrome, Leigh syndrome, Angelman syndrome, Pader-Willi syndrome, Smith-Lemli-Opitx syndrome, Kabuki syndrome, lactic acidosis, stroke-like episodes (MELAS), attention deficit hyperactivity disorder (ADHD), and intellectual disabilities. These findings underscore the importance of elucidating the molecular mechanisms underlying mitochondrial impairments in neurodevelopmental disorders, offering new avenues for early diagnosis and personalized treatment approaches. By unraveling the complex interplay between mitochondrial biology and neurodevelopment, healthcare practitioners can pave the way for tailored interventions aimed at optimizing outcomes and enhancing the quality of life for individuals affected by ASD and concomitant mitochondrial dysfunction.
Primary Mitochondrial Disease (PMD) vs Secondary Mitochondrial Dysfunction (SMD)
From a scientific standpoint, mitochondrial dysfunction encompasses a diverse array of conditions characterized by impaired cellular energy production due to abnormal oxidative phosphorylation (OXPHOS). Two broad categories used to classify mitochondrial dysfunction are Primary Mitochondrial Disease (PMD) and Secondary Mitochondrial Dysfunction (SMD).
PMD refers to a group of genetic disorders that result from mutations in the genes of either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA) that directly affect mitochondrial function. These mutations can disrupt various aspects of mitochondrial function, including energy production, regulation of reactive oxygen species (ROS), and mitochondrial dynamics. PMD can manifest in a wide range of symptoms affecting multiple organ systems, including the brain, muscles, heart, and other vital organs. Examples of PMD include Leigh syndrome, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), and mitochondrial myopathies.
SMD, on the other hand, refers to mitochondrial dysfunction that arises as a consequence of other underlying conditions or factors, rather than directly from genetic mutations affecting mitochondrial function. These underlying conditions can include environmental toxins, infections, metabolic disorders, and neurodegenerative diseases, among others. SMD can occur when these conditions disrupt cellular processes that are essential for maintaining mitochondrial health, such as oxidative phosphorylation, mitochondrial dynamics, or mitochondrial quality control mechanisms. As a result, SMD can contribute to a wide range of health problems, including neurodegenerative diseases, cardiovascular disorders, metabolic syndrome, and aging-related conditions.
SMD has been observed in a wide range of conditions, including autoimmune disorders like multiple sclerosis and lupus (1,2,3 ), suggesting a complex interplay between mitochondrial dysfunction and immune dysregulation. Additionally, aging is recognized as a significant contributor to oxidative stress throughout the body, heightening the susceptibility to SMD.
Altered dynamics of mitochondrial fusion and fission have emerged as a common feature in neurodegenerative diseases such as Alzheimer's and Parkinson's, underscoring the role of mitochondrial dysfunction in neurodegeneration. Furthermore, many metabolic disorders encompass SMD within their phenotypic spectrum.
In clinical contexts, abnormal biomarkers indicative of mitochondrial function are frequently detected in individuals with autism, suggesting a potential link between mitochondrial dysfunction and neurodevelopmental disorders. Depending on the dataset considered, SMD may be present in a notable subset, or even the majority, of cases.
While genetic testing ideally serves to distinguish between PMD and SMD, facilitating tailored treatments and prognoses, the overlap in clinical presentations can complicate differentiation. Nevertheless, standard treatments for PMD have demonstrated efficacy in managing SMD, highlighting the therapeutic potential of targeting mitochondrial dysfunction across various conditions.
Genetic Testing: Pathogenic Variants in Mitochondrial and Inflammation-Associated Genes
In the realm of mitochondrial dysfunction, there exists a fascinating yet complex interplay between genetic variations and disease manifestation.
One such example is the presence of pathogenic variants in genes encoding proteins crucial for mitochondrial homeostasis and function (e.g., POLG, MFN2, OPA1, ATP5A1, SDHB, ACAD9, TWNK, HSPA9, TRIT1)(4,5). These variants may alter the structure or function of mitochondrial proteins, leading to impaired mitochondrial processes such as oxidative phosphorylation, mitochondrial dynamics, or mitochondrial protein import. Consequently, even in the absence of a clear link to established mitochondrial diseases, these mutations can exert detrimental effects on cellular energetics and metabolism.
Moreover, pathogenic variants in nuclear genes encoding proteins involved in mitochondrial biogenesis, maintenance, or quality control mechanisms can also disrupt mitochondrial function (e.g., TWINKLE, TWNKIP1, POLG2, POLG3, TFAM, GFM1, MPV17, MGME1). For instance, mutations in genes regulating mitochondrial DNA replication, repair, or segregation may compromise the integrity of the mitochondrial genome, leading to mitochondrial dysfunction and cellular stress. Similarly, mutations affecting mitochondrial protein import machinery or mitochondrial proteases can disrupt the proper functioning of mitochondrial proteins, further exacerbating mitochondrial dysfunction.
While numerous pathogenic variant mutations have been identified, not all are directly associated with known mitochondrial disorders. Nonetheless, these mutations have the potential to disrupt mitochondrial function and contribute to cellular dysfunction, albeit through mechanisms that are not yet fully understood.
Think of a cell like a big building with lots of rooms, each room doing different jobs to keep the building running smoothly. In each room, there are small circuit boards called mitochondria. These circuit boards are like tiny power stations, providing energy for the room to do its job.
Now, imagine some of these circuit boards are working perfectly fine, shining brightly and providing lots of energy. But others are damaged or broken, flickering on and off, struggling to keep up. These damaged circuit boards represent dysfunctional mitochondria—they can't produce enough energy for the room to function properly.
In this way, dysfunctional mitochondria are like broken parts in a machine. They disrupt the flow of energy, making it harder for the cell to do its job. Just like how a broken circuit board can cause problems in a building, dysfunctional mitochondria can cause issues in a cell, affecting its overall health and function.

Understanding the impact of these pathogenic variants on mitochondrial function is essential for unraveling the complexity of mitochondrial biology and its role in health and disease. While the clinical consequences of these mutations may not always align with classical mitochondrial disorders, their contribution to mitochondrial dysfunction underscores the need for comprehensive genetic analyses and functional studies (6). By elucidating the molecular mechanisms underlying these pathogenic variants, researchers can identify novel therapeutic targets and develop personalized treatment strategies for individuals affected by mitochondrial dysfunction. Thus, exploring the diverse landscape of pathogenic variant mutations offers valuable insights into the broader spectrum of mitochondrial diseases and paves the way for advancements in precision medicine approaches targeting mitochondrial dysfunction.
A pathogenic but nonsense mutation?
As the field of genetics rapidly advances, genetic testing has become an invaluable tool for understanding potential health risks and inherited conditions in children. However, it's crucial to recognize that genetic test reports provide only a snapshot of your child's genetic makeup at a specific moment in time.
Our understanding of genetics is constantly evolving, with new discoveries and advancements occurring regularly. What may be considered a benign or nonsensical finding today could potentially be identified as pathogenic in the future. Therefore, it's essential not to view genetic test results as definitive and unchanging.
I would also encourage you to approach genetic testing as part of an ongoing dialogue with healthcare professionals. Regular re-evaluation and rechecking of genetic findings are critical to ensure that your child's healthcare plan remains up-to-date and informed by the latest scientific knowledge.
A negative genetic result doesn't necessarily mean your child is not affected by potentially pathogenic variants. Genetic testing may not detect all genetic mutations or variations associated with a particular condition. Moreover, some conditions may have complex or multifactorial causes beyond genetic factors alone. It's also important to note that the interpretation of genetic data can be challenging and may vary in quality. Identifying clinically relevant genetic variants requires expertise in bioinformatics and careful assessment of the data. In some cases, variants may be missed or misinterpreted, leading to potential limitations in diagnostic accuracy and clinical utility.
By staying informed about advancements in genetics and participating in discussions with your healthcare provider, you can remain proactive in monitoring your child's health. Furthermore, by checking resources like ClinVar, one can stay informed about ongoing changes in genetic data, ensuring they have the most up-to-date information on genetic variants and their clinical significance. Remember, genetic testing is just one piece of the puzzle, and ongoing evaluation and re-evaluation of findings are essential to provide the best possible care for your child.
https://www.ncbi.nlm.nih.gov/clinvar/
https://www.clinicalgenome.org/
https://clinvarminer.genetics.utah.edu/
Oxidative Phosphorylation
Oxidative phosphorylation (OXPHOS) is a crucial process that occurs within cells, particularly in the mitochondria, the cell's powerhouses. It is part of cellular respiration, the process by which cells convert nutrients into energy in the form of adenosine triphosphate (ATP), which is used to power various cellular activities.
During oxidative phosphorylation, enzymes within the mitochondria use oxygen to help convert energy stored in the form of glucose and other nutrients into ATP. This process involves a series of biochemical reactions, primarily carried out by protein complexes located within the inner mitochondrial membrane.
Imagine your body as a busy factory, and the mitochondria are like power plants within this factory. These power plants, called mitochondria, work tirelessly to produce energy for the cells to function properly.
Think of glucose, a type of sugar found in food, as the fuel that powers these mitochondria. Just like a power plant burns fuel to generate electricity, mitochondria use glucose to produce energy in the form of ATP (adenosine triphosphate).
Now, let's break down how this process works:
.jpg)
-
Glucose enters the mitochondria and undergoes a series of chemical reactions, similar to gears turning in a machine. These reactions release energy stored in glucose molecules.
-
As glucose is broken down, electrons are passed along a chain of special molecules, like a relay race. This chain is called the electron transport chain.
-
As the electrons move along the chain, they release energy, which is used to pump protons (like tiny energy packets) across the mitochondrial membrane.
-
The buildup of protons creates a sort of energy reservoir, like water stored behind a dam. When the protons flow back across the membrane through a special channel, they release energy, just like water flowing through a hydroelectric dam.
-
This released energy is used to make ATP, which serves as the cell's energy currency. It's like the batteries that power all the machinery in the factory.
So, in simple terms, oxidative phosphorylation is like a power plant in your cells that converts glucose into ATP, the energy source that keeps your body running smoothly. Just like a well-oiled machine, your mitochondria work tirelessly to produce the energy your cells need to function properly.
While oxidative phosphorylation plays a crucial role in metabolism, it generates reactive oxygen species (ROS) such as superoxide and hydrogen peroxide, leading to the production of free radicals. This process can damage cells and contribute to the development of various diseases. The enzymes involved in oxidative phosphorylation are commonly referred to as complex I, complex II, complex III, complex IV, and complex V.
Within the mitochondria, the conversion of one molecule of glucose into carbon dioxide and water can yield up to 36 ATP molecules. However, this process relies on the presence of oxygen, and in its absence, a less efficient pathway is utilized. Some athletes attempt to enhance their oxygen levels to gain an advantage in sports.
Adenosine triphosphate (ATP) serves as a vital molecule in cells, acting as a coenzyme. Often described as the "molecular unit of currency" for intracellular energy transfer, ATP transports chemical energy within cells to support various metabolic processes. These processes include protein synthesis, membrane synthesis, cellular movement, division, and solute transport.
When ATP is broken down into adenosine diphosphate (ADP) and phosphate (Pi), energy is released. By analyzing the levels of specific byproducts produced during the five major steps between glucose and ATP synthesis, it is possible to identify deficiencies in the five enzyme complexes involved in oxidative phosphorylation.
Studies confirm that ASD is associated with unique changes in mitochondrial metabolism, including elevated respiration rates and morphological alterations, where alterations in mitochondrial morphology were associated with both ETC Complex I+III and IV activity as well as the difference between ETC Complex I+III and IV activity (7,8,9,10). However, unraveling the intricacies of complex I-IV deficiency can be challenging, as it involves a complex interplay of multiple genes encoding various components of the complex.
For example, complex I deficiency is associated with mutations in at least 51 different genes, each encoding crucial components of the complex. Down-regulation or mutations in any of these genes can disrupt the assembly or function of complex I, impairing its ability to carry out its role in oxidative phosphorylation (11). This complexity underscores the need for comprehensive genetic analyses and molecular investigations to pinpoint the exact genetic cause of complex I-IV deficiency and guide targeted treatment strategies.
References:
-
Barrera, M.-J., Aguilera, S., Castro, I., Carvajal, et al. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: Potential role in Sjögren’s syndrome. (2021) Autoimmunity Reviews, 20(8), 102867. https://doi.org/10.1016/j.autrev.2021.102867
-
Hanaford, A., Johnson, S.C. The immune system as a driver of mitochondrial disease pathogenesis: a review of evidence. (2022) Orphanet J Rare Dis 17, 335. https://doi.org/10.1186/s13023-022-02495-3
-
Quintero-González, D. C., Muñoz-Urbano, M., & Vásquez, G. (2022). Mitochondria as a key player in systemic lupus erythematosus. Autoimmunity, 55(8), 497–505. https://doi.org/10.1080/08916934.2022.2112181
-
Rahman, J. Novel Diagnostic and Therapeutic Approaches for Mitochondrial Disorders. (2020) Genetics and Genomic MedicineUCL Great Ormond Street Institute of Child Health, London. Retrieved from https://discovery.ucl.ac.uk/id/eprint/10103427/8/Rahman_10103427_thesis_redacted.pdf
-
Saneto, R. Mitochondrial diseases: expanding the diagnosis in the era of genetic testing. (2020) J Transl Genet Genom. 4:384-428. doi: 10.20517/jtgg.2020.40
-
Wortmann, S., Oud, M., Alders, M., Coene, K., Van Der Crabben, et al. How to proceed after “negative” exome: A review on genetic diagnostics, limitations, challenges, and emerging new multiomics techniques. (2022) Journal of Inherited Metabolic Disease, 45(4), 663–681. https://doi.org/10.1002/jimd.12507
-
Frye, R., Lionnard, L., Singh, I. et al. Mitochondrial morphology is associated with respiratory chain uncoupling in autism spectrum disorder. (2021) Transl Psychiatry 11, 527. https://doi.org/10.1038/s41398-021-01647-6
-
Nunes, T., Latini, A., Gaspar, J. Mitochondrial Dysfunction in Autism Spectrum Disorders. (2023) In: El Idrissi, A., McCloskey, D. (eds) Neurobiology of Autism Spectrum Disorders. Springer, Cham. https://doi.org/10.1007/978-3-031-42383-3_5
-
Delhey, L.; Nur Kilinc, E.; Yin, L.; Slattery, J.; Tippett, M.; Rose, S.; Bennuri, S.; Kahler, S.; et al. The Effect of Mitochondrial Supplements on Mitochondrial Activity in Children with Autism Spectrum Disorder. J. Clin. Med. 2017, 6, 18. https://doi.org/10.3390/jcm6020018
-
Gonçalves, F., Alves, C., Heuer, B., Peterson, J., Viaene, A. N.,et al. Primary Mitochondrial Disorders of the Pediatric Central Nervous System: Neuroimaging Findings. (2020) Radiographics, 40(7), 2042–2067. https://doi.org/10.1148/rg.2020200052
-
Lloyd-Thomas, P. Mitochondrial Disease and Autsim. (2017) Retrieved from https://www.epiphanyasd.com/2017/02/mitochondrial-disease-and-autsim.html